
1-(BROMOMETHYL)-3-PHENOXYBENZENE synthesis
- Product Name:1-(BROMOMETHYL)-3-PHENOXYBENZENE
- CAS Number:51632-16-7
- Molecular formula:C13H11BrO
- Molecular Weight:263.13
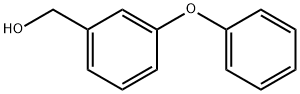
13826-35-2
51632-16-7
General procedure for the synthesis of m-phenoxybenzyl bromide from 3-phenoxybenzyl alcohol: 3-phenoxybenzyl alcohol (2.09 g, 10.0 mmol) was dissolved in 18.7 mL of anhydrous dichloromethane and cooled to 0 °C. Subsequently, a solution of phosphorus tribromide (0.35 mL, 3.80 mmol) in 4.7 mL of dichloromethane was slowly added. The reaction mixture was naturally warmed to room temperature with continuous stirring for 30 minutes. Upon completion of the reaction, the reaction was quenched with saturated aqueous sodium bicarbonate and extracted with ether. The organic phases were combined, dried with anhydrous magnesium sulfate and concentrated under reduced pressure. Finally, purification by fast chromatography (eluent: hexane/ethyl acetate, 8:2) afforded 1.98 g of the target product, m-phenoxybenzyl bromide, as a colorless oil in 72% yield. The spectral data of the product were in agreement with those reported in the literature (Surman, MD; Mulvihill, MJ J. Org. Chem. 2002, 67, 4115-4121).
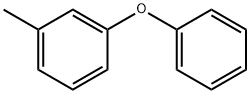
3586-14-9
211 suppliers
$9.00/5g
51632-16-7
97 suppliers
$74.23/250mgs:
Yield: 86%
Reaction Conditions:
with N-Bromosuccinimide;2,2'-azobis(isobutyronitrile) in tetrachloromethane for 3 h;Reflux;
Steps:
5.2.13. 1-(Azidomethyl)-3-phenoxybenzene (13)
A solution of m-(phenoxy)toluene (3.68 g; 20 mmol) in 45 mL CCl4 was treated with N-bromosuccinimide (5.34 g; 30 mmol) and azo-bis(isobutyronitrile) (15 mg) and the reaction mixture was refluxed for 3 h. The solution was allowed to cool to room temperature and then placed in an ice bath. A white precipitate formed which was filtered off and the filtrate was evaporated, leaving pure compound 1 as a yellow oil (4.5 g; 86% yield). 1H NMR (CDCl3): δ 4.4 (s, 2H), 6.9-7.41 (m, 9H). This was used in the next step. To sodium azide (3.25 g; 50 mmol) in dry DMSO (80 mL) was added compound 12 (4.5 g; 17.1 mmol) and the reaction mixture was stirred overnight at room temperature. The reaction mixture was cooled in an ice bath and quenched with water (50 mL). The aqueous layer was extracted with ethyl ether (3 × 30 mL). The combined ethyl ether extracts were washed with water (2 × 20 mL) and dried over anhydrous sodium sulfate. The solvent was removed, leaving a crude product which was purified using flash chromatography (silica gel/methylene chloride/hexanes) to give compound 13 as a colorless oil (3.31 g; 60% yield). 1H NMR (CDCl3): δ 4.25 (s, 2H), 6.07-7.45 (m, 9H).
References:
Dou, Dengfeng;Mandadapu, Sivakoteswara Rao;Alliston, Kevin R.;Kim, Yunjeong;Chang, Kyeong-Ok;Groutas, William C. [Bioorganic and Medicinal Chemistry,2011,vol. 19,# 19,p. 5749 - 5755] Location in patent:experimental part
13826-35-2
330 suppliers
$6.00/5g
51632-16-7
97 suppliers
$74.23/250mgs:
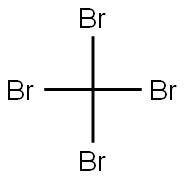
558-13-4
265 suppliers
$10.00/1g
13826-35-2
330 suppliers
$6.00/5g
51632-16-7
97 suppliers
$74.23/250mgs:
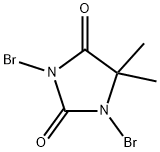
77-48-5
558 suppliers
$11.00/10g
3586-14-9
211 suppliers
$9.00/5g
51632-16-7
97 suppliers
$74.23/250mgs:
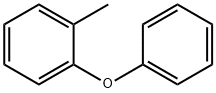
3991-61-5
1 suppliers
inquiry
3586-14-9
211 suppliers
$9.00/5g
51632-16-7
97 suppliers
$74.23/250mgs:
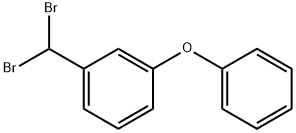
53874-67-2
1 suppliers
inquiry