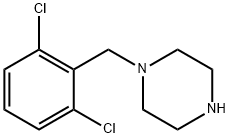
1-(2,6-DICHLOROBENZYL)PIPERAZINE synthesis
- Product Name:1-(2,6-DICHLOROBENZYL)PIPERAZINE
- CAS Number:102292-50-2
- Molecular formula:C11H14Cl2N2
- Molecular Weight:245.15

110-85-0
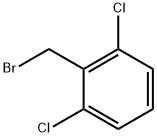
20443-98-5
102292-50-2
1. Dissolve piperazine (112 mmol, 6.0 eq.) in THF (180 mL) at 0 °C to prepare a solution. 2. 2,6-dichlorobenzyl bromide (4.5 g, 18.8 mmol) was dissolved in THF (20 mL) to prepare a solution and slowly added dropwise to the THF solution of piperazine over 10 minutes. 3. After the dropwise addition was completed, the reaction mixture was slowly warmed to room temperature and stirred continuously for 24 hours. 4. Upon completion of the reaction, the THF solvent was removed by distillation under reduced pressure. 5. The crude product was resuspended in a mixture of dichloromethane (DCM) and water and extracted twice with DCM. 6. The organic layers were combined, dried over anhydrous sodium sulfate (Na2SO4), filtered and concentrated. 7. The crude product was purified using combiflash fast chromatography eluting with a gradient of 0 to 20% methanol in DCM to afford the target compound 1-(2,6-dichlorobenzyl)piperazine (2.3 g, 50% yield). 8. The structure of the product was confirmed by 1H NMR, 13C NMR and HRMS. 1H NMR (400 MHz, methanol-d4) δ 7.62-7.30 (m, 2H), 7.23 (dd, J = 8.7, 7.4 Hz, 1H), 3.74 (s, 2H), 2.92-2.69 (m, 4H), 2.56 (t, J = 4.9 Hz, 4H). 13C NMR (101 MHz, MeOD) δ 136.76, 133.67, 129.18, 128.24, 56.55, 53.41, 44.95. HRMS (m/z): [M+] calculated value C11H14Cl2N2 245.15, measured value 245.06.
110-85-0
584 suppliers
$5.00/5 g

2014-83-7
297 suppliers
$9.00/5g
102292-50-2
88 suppliers
$16.00/250mg
Yield: 95.7%
Reaction Conditions:
in tetrahydrofuran for 4 h;Reflux;
Steps:
5.1.3. General procedure for the synthesis of various substituted benzylpiperazines (3a-s)
General procedure: Anhydrous piperazine (6.89 g, 80 mmol) was dissolved in 40 mL of freshly distilled THF. Once the piperazine was fully dissolved, 2.303 mL (20 mmol) of benzyl chloride was added dropwise. The reaction mixture was then refluxed for about 4 h until benzyl chloride disappeared, as assessed by TLC. The stirring mixture was allowed to cool, and then filtered. The filtrate was concentrated in a rotary evaporator and then diluted with EtOAc (100 mL) and water (50 mL), which was then made basic (pH>12) with a saturated 1 N NaOH aqueous solution and separated. The organic phase was washed with water (4 × 100 mL), brine (2 × 100 mL), dried over Na2SO4 and concentrated to yield an oil. Column chromatography (PE/EtOAc = 1:1 to EtOAc/MeOH = 5:1) afforded a pale yellow liquid.
References:
Zhang, Cunlong;Tan, Chunyan;Zu, Xuyu;Zhai, Xin;Liu, Feng;Chu, Bizhu;Ma, Xiaohua;Chen, Yuzong;Gong, Ping;Jiang, Yuyang [European Journal of Medicinal Chemistry,2011,vol. 46,# 4,p. 1404 - 1414] Location in patent:experimental part
110-85-0
584 suppliers
$5.00/5 g
20443-98-5
213 suppliers
$8.00/5g
102292-50-2
88 suppliers
$16.00/250mg
20443-98-5
213 suppliers
$8.00/5g
102292-50-2
88 suppliers
$16.00/250mg